
Le terme de « tumeur primitive du pancréas» s’emploie pour décrire une tumeur dont l’origine est une cellule pancréatique. En fait, ce terme regroupe un grand nombre de tumeurs au comportement et au pronostic variés, que l’on peut séparer en deux groupes bien distincts en fonction des cellules d’origine en tumeurs exocrines ou endocrines.
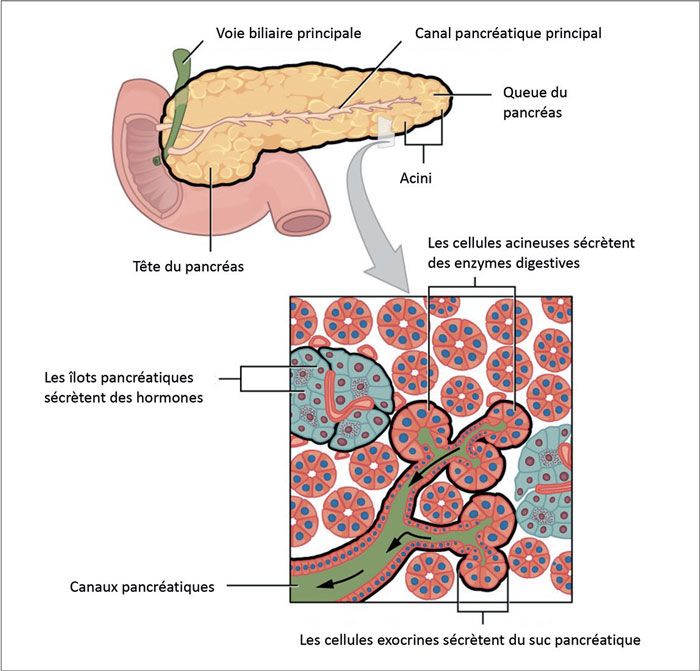
Figure 1. L’origine exocrine ou endocrine des tumeurs.
Les tumeurs exocrines
L’adénocarcinome du pancréas (AP) est la forme histologique largement prédominante du cancer du pancréas. Il s’agit d’une tumeur développée à partir des cellules épithéliales des canaux pancréatiques (adénocarcinome canalaire). L’AP correspond à 90 % des tumeurs du pancréas[3] et devrait être la 2e cause de mortalité par cancer en Europe et aux États Unis en 2040[3,11]. En France, son incidence a doublé chez les hommes et triplé chez les femmes entre 1982 et 2012[12] et environ 15000 nouveaux cas d’AP sont diagnostiqués tous les ans. L’âge moyen au diagnostic est de 73 ans chez la femme et de 68 ans chez l’homme[1].
L’AP est rapidement évolutif, avec une extension locale et métastatique par voies lymphatique, nerveuse et sanguine[13]. Dans 2/3 des cas, la lésion apparaît à la tête du pancréas. Il reste le cancer digestif dont le pronostic est le plus défavorable, avec un taux de survie globale (SG) de 5 % à 5 ans, tous stades confondus[14]. Les cancers non canalaires sont hétérogènes et plus rares :
• le carcinome à cellules acineuses, développé à partir des acini pancréatiques et représentant moins de 1 % des tumeurs du pancréas exocrine ;
• le carcinome adénosquameux ;
• le carcinome colloïde ;
• le carcinome hépatoïde ;
• de même que des tumeurs conjonctives, lymphomateuses, ou encore le pancréatoblastome[15,16].
Les tumeurs neuroendocrines
Les tumeurs neuroendocrines pancréatiques (TNEP) sont un groupe hétérogène de tumeurs rares[17]. Historiquement, elles représentent 23 % des néoplasies pancréatiques mais leur incidence semble être en augmentation (jusqu’à 10 %)[18-20]. Leur croissance tumorale, le plus souvent relativement lente, permet des taux de survie prolongés même aux stades métastatiques[17]. Les TNEP sont le plus souvent sporadiques et diagnostiquées fortuitement ou devant des signes cliniques aspécifiques en lien avec les localisations tumorales. Moins de 20 % des TNEP sont fonctionnelles et sont responsables de symptômes liés à l’hypersécrétion d’une hormone devant être traités en priorité. Les principaux facteurs pronostiques sont :
• la différenciation tumorale et le grade histologique ;
• le stade tumoral ;
• la pente évolutive ;
• et le volume tumoral, notamment hépatique[19,21].
Le traitement des TNEP localisées repose sur la résection chirurgicale, à l’exception de certains incidentalomes sporadiques < 2 cm[18,21].
Avec le soutien institutionnel de
